
Latest Content by PharmaCompass

 KEY PRODUCTS KEY SERVICES
KEY PRODUCTS KEY SERVICES
European CDMO and Gx manufacturer with 75 years of experience in delivering premium APIs to pharmaceutical partners worldwide.
About
CPhI North AmericaCPhI North America
Industry Trade Show
Attending
02-04 June, 2026
Industry Trade Show
Booth #E2C71
16-18 June, 2026
BIO International Conv...BIO International Convention
Industry Trade Show
Attending
22-25 June, 2026
CONTACT DETAILS



Digital content ![]()
INTERVIEW #SpeakPharma
VLOG #PharmaReel

CORPORATE CONTENT #SupplierSpotlight
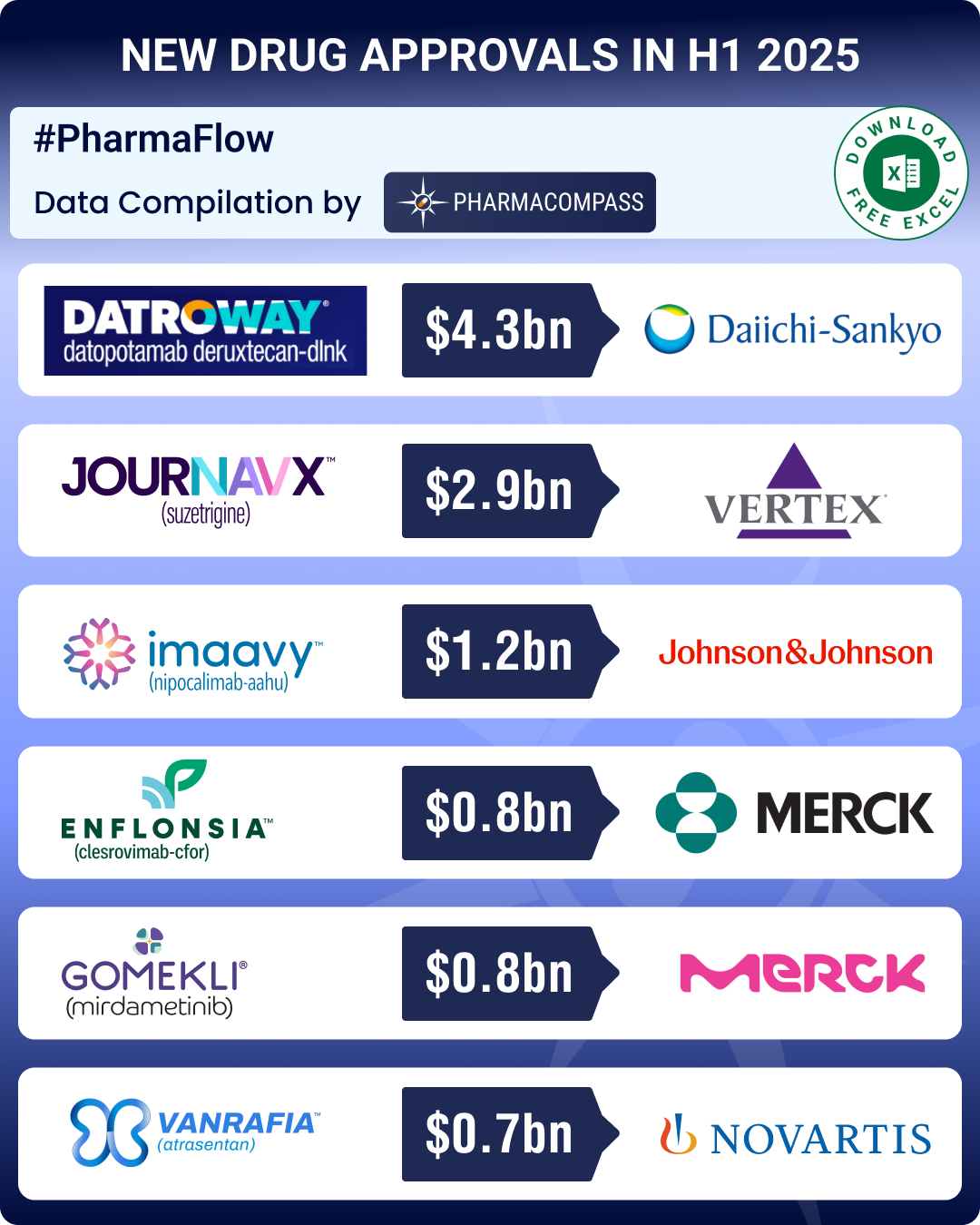
DATA COMPILATION #PharmaFlow

NEWS #PharmaBuzz
24 Oct 2025
// PRESS RELEASE
25 Jun 2025
// PRESS RELEASE
25 Jun 2025
// PRESS RELEASE
14 May 2025
// PRESS RELEASE
01 Jan 2025
// PRESS RELEASE
10 Oct 2024
// PRESS RELEASE
USDMF
 FULL SCREEN VIEW Click here to open all results in a new tab [this preview display 10 results]
FULL SCREEN VIEW Click here to open all results in a new tab [this preview display 10 results]Drugs in Development
FULL SCREEN VIEW Click here to open all results in a new tab [this preview display 10 results]ALL Intermediates
FULL SCREEN VIEW Click here to open all results in a new tab [this preview display 10 results]KEY PRODUCTS
TOP RANKED SUPPLIER FOR:























Scale Up
FULL SCREEN VIEW Click here to open all results in a new tab [this preview display 10 results]Halogenation
FULL SCREEN VIEW Click here to open all results in a new tab [this preview display 10 results]Phosgenation
FULL SCREEN VIEW Click here to open all results in a new tab [this preview display 10 results]Hydrogenation
FULL SCREEN VIEW Click here to open all results in a new tab [this preview display 10 results]Micronization
FULL SCREEN VIEW Click here to open all results in a new tab [this preview display 10 results]Small Molecules
FULL SCREEN VIEW Click here to open all results in a new tab [this preview display 10 results]GMP Manufacturing
FULL SCREEN VIEW Click here to open all results in a new tab [this preview display 10 results]Hazardous Chemistry
FULL SCREEN VIEW Click here to open all results in a new tab [this preview display 10 results]High Potency APIs (HPAPIs)
FULL SCREEN VIEW Click here to open all results in a new tab [this preview display 10 results]Oligonucleotide / Polynucleotide
FULL SCREEN VIEW Click here to open all results in a new tab [this preview display 10 results]High Pressure Reaction (> 100 psi)
FULL SCREEN VIEW Click here to open all results in a new tab [this preview display 10 results]Process Development & Optimization
FULL SCREEN VIEW Click here to open all results in a new tab [this preview display 10 results]Low Temperature / Cryogenic conditions
FULL SCREEN VIEW Click here to open all results in a new tab [this preview display 10 results]Custom Synthesis & Manufacturing
FULL SCREEN VIEW Click here to open all results in a new tab [this preview display 10 results]KEY SERVICES
TOP RANKED SUPPLIER FOR:





GDUFA
FULL SCREEN VIEW Click here to open all results in a new tab [this preview display 10 results]THIRD-PARTY AUDIT

Country : Poland
City/Region : Sieradz
Audit Date : 2025-10-14
Audit Type : On-Site


Country : Poland
City/Region : Starogard Gdański
Audit Date : Nov-25
Audit Type : On-Site

 FULL SCREEN VIEW Click here to open all results in a new tab [this preview display 10 results]
FULL SCREEN VIEW Click here to open all results in a new tab [this preview display 10 results]ABOUT THIS PAGE
Polpharma is a supplier offers 77 products (APIs, Excipients or Intermediates).
Find Alendronate Sodium bulk with DMF, CEP, JDMF offered by Polpharma
Find Hydrochlorothiazide bulk with DMF, CEP, JDMF offered by Polpharma
Find Ibandronate Sodium bulk with DMF, CEP, JDMF offered by Polpharma
Find Lamotrigine bulk with DMF, CEP, JDMF offered by Polpharma
Find Risedronate Sodium bulk with DMF, CEP, JDMF offered by Polpharma
Find Sildenafil Citrate bulk with DMF, CEP, JDMF offered by Polpharma
Find Tadalafil bulk with DMF, CEP, JDMF offered by Polpharma
Find Vardenafil Hydrochloride bulk with DMF, CEP, JDMF offered by Polpharma
Find Zoledronic Acid bulk with DMF, CEP, JDMF offered by Polpharma
Find Acetazolamide bulk with DMF, JDMF offered by Polpharma
Find Baclofen bulk with DMF, JDMF offered by Polpharma
Find Carbamazepine bulk with DMF, CEP offered by Polpharma
Find Dapagliflozin bulk with DMF, JDMF offered by Polpharma
Find Etodolac bulk with DMF, JDMF offered by Polpharma
Find Pentoxifylline bulk with DMF, CEP offered by Polpharma
Find Sodium Salicylate bulk with DMF, JDMF offered by Polpharma
Find Abemaciclib bulk with DMF offered by Polpharma
Find Apixaban bulk with DMF offered by Polpharma
Find Apremilast bulk with DMF offered by Polpharma
Find Aripiprazole bulk with DMF offered by Polpharma
Find Baricitinib bulk with DMF offered by Polpharma
Find Brexpiprazole bulk with DMF offered by Polpharma
Find Carbamazepine bulk with CEP offered by Polpharma
Find Carvedilol bulk with DMF offered by Polpharma
Find Carvedilol Phosphate bulk with DMF offered by Polpharma
Find Dapagliflozin Propanediol Monohydrate bulk with DMF offered by Polpharma
Find Empagliflozin bulk with DMF offered by Polpharma
Find Enzalutamide bulk with DMF offered by Polpharma
Find Ibandronate Sodium bulk with CEP offered by Polpharma
Find Isavuconazonium Sulfate bulk with DMF offered by Polpharma
Find Lamotrigine bulk with JDMF offered by Polpharma
Find Molsidomine bulk with CEP offered by Polpharma
Find Palbociclib bulk with DMF offered by Polpharma
Find Pimavanserin Tartrate bulk with DMF offered by Polpharma
Find Piracetam bulk with CEP offered by Polpharma
Find Rivaroxaban bulk with DMF offered by Polpharma
Find Sacubitril-Valsartan bulk with DMF offered by Polpharma
Find Safinamide Methanesulfonate bulk with DMF offered by Polpharma
Find Sildenafil Citrate bulk with CEP offered by Polpharma
Find Sitagliptin Hydrochloride bulk with DMF offered by Polpharma
Find Ticagrelor bulk with DMF offered by Polpharma
Find Tolterodine Tartrate bulk with CEP offered by Polpharma
Find Topiramate bulk with DMF offered by Polpharma
Find Trametinib bulk with DMF offered by Polpharma
Find Xylometazoline Hydrochloride bulk with CEP offered by Polpharma
Find Acenocoumarol bulk offered by Polpharma
Find Acetazolamide bulk offered by Polpharma
Find Alendronate Sodium bulk offered by Polpharma
Find Aniracetam bulk offered by Polpharma
Find Antazoline HCl bulk offered by Polpharma
Find Antazoline Mesylate bulk offered by Polpharma
Find Antazoline Sulfate bulk offered by Polpharma
Find Apixaban bulk offered by Polpharma
Find Aripiprazole bulk offered by Polpharma
Find Aripiprazole Lauroxil bulk offered by Polpharma
Find Baclofen bulk offered by Polpharma
Find Carbamazepine bulk offered by Polpharma
Find Carvedilol bulk offered by Polpharma
Find Carvedilol Phosphate bulk offered by Polpharma
Find CAS 66376-36-1 bulk offered by Polpharma
Find Clemastine bulk offered by Polpharma
Find Clopamide bulk offered by Polpharma
Find Denotivir bulk offered by Polpharma
Find Disulfiram bulk offered by Polpharma
Find Etodolac bulk offered by Polpharma
Find Hydrochlorothiazide bulk offered by Polpharma
Find Nefopam Hydrochloride bulk offered by Polpharma
Find Opipramol Dihydrochloride bulk offered by Polpharma
Find Phenyl Salicylate bulk offered by Polpharma
Find Risdiplam bulk offered by Polpharma
Find Risedronate Sodium bulk offered by Polpharma
Find Rivaroxaban bulk offered by Polpharma
Find Salicylamide bulk offered by Polpharma
Find Talazoparib bulk offered by Polpharma
Find Ticagrelor bulk offered by Polpharma
Find Topiramate bulk offered by Polpharma
Find Vismodegib bulk offered by Polpharma

 Polpharma
Polpharma



















 Eurofins Assurance: Ensuring compliance, quality, and safety with expert inspection and certification.
Eurofins Assurance: Ensuring compliance, quality, and safety with expert inspection and certification.