

1. Arn-509
2. Erleada
1. Arn-509
2. 956104-40-8
3. Erleada
4. Jnj-56021927
5. Arn 509
6. Apalutamide [inn]
7. Arn509
8. Apalutamide (arn-509)
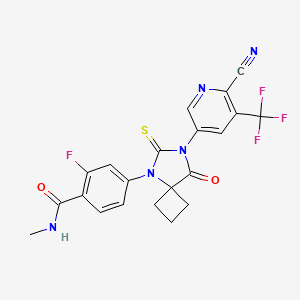
9. 4-(7-(6-cyano-5-(trifluoromethyl)pyridin-3-yl)-8-oxo-6-thioxo-5,7-diazaspiro[3.4]octan-5-yl)-2-fluoro-n-methylbenzamide
10. 4t36h88ua7
11. 956104-40-8 (free Base)
12. 4-[7-[6-cyano-5-(trifluoromethyl)pyridin-3-yl]-8-oxo-6-sulfanylidene-5,7-diazaspiro[3.4]octan-5-yl]-2-fluoro-n-methylbenzamide
13. 4-{7-[6-cyano-5-(trifluoromethyl)pyridin-3-yl]-8-oxo-6-sulfanylidene-5,7-diazaspiro[3.4]octan-5-yl}-2-fluoro-n-methylbenzamide
14. 4-(7-(6-cyano-5-(trifluoromethyl)pyridin-3-yl)-8-oxo-6-thioxo-5,7-diazaspiro(3.4)octan-5-yl)-2-fluoro-n-methylbenzamide
15. Unii-4t36h88ua7
16. Ar509
17. Apalutamidearn509
18. Erleada (tn)
19. Jnj 56021927
20. Apalutamide (jan/inn)
21. Apalutamide [mi]
22. Apalutamide [jan]
23. Apalutamide [who-dd]
24. Mls006011109
25. Schembl909297
26. Gtpl9043
27. Chembl3183409
28. Apalutamide [orange Book]
29. Dtxsid40241899
30. Ex-a089
31. Hms3656n12
32. Amy24182
33. Bcp05829
34. Ar509/ar-509
35. Bdbm50094975
36. Mfcd22380626
37. Nsc771649
38. Nsc794776
39. S2840
40. Zinc43174901
41. Akos025401932
42. Ccg-264760
43. Cs-0885
44. Db11901
45. Nsc-771649
46. Nsc-794776
47. Pb27306
48. Ncgc00346725-01
49. Ncgc00346725-02
50. Ncgc00346725-06
51. Ac-27403
52. As-35181
53. Hy-16060
54. Smr004702891
55. Sw220300-1
56. 24872560, Erleada, C21h15f4n5o2s
57. D11040
58. J-519596
59. Q21098975
60. Benzamide, 4-(7-(6-cyano-5-(trifluoromethyl)-3-pyridinyl)-8-oxo-6-thioxo-5,7-diazaspiro(3.4)oct-5-yl)-2-fluoro-n-methyl-
| Molecular Weight | 477.4 g/mol |
|---|---|
| Molecular Formula | C21H15F4N5O2S |
| XLogP3 | 3 |
| Hydrogen Bond Donor Count | 1 |
| Hydrogen Bond Acceptor Count | 9 |
| Rotatable Bond Count | 3 |
| Exact Mass | 477.08825856 g/mol |
| Monoisotopic Mass | 477.08825856 g/mol |
| Topological Polar Surface Area | 121 Ų |
| Heavy Atom Count | 33 |
| Formal Charge | 0 |
| Complexity | 886 |
| Isotope Atom Count | 0 |
| Defined Atom Stereocenter Count | 0 |
| Undefined Atom Stereocenter Count | 0 |
| Defined Bond Stereocenter Count | 0 |
| Undefined Bond Stereocenter Count | 0 |
| Covalently Bonded Unit Count | 1 |
Indicated for the treatment of patients with non-metastatic, castration-resistant prostate cancer (NM-CRPC).
FDA Label
Erleada is indicated:
in adult men for the treatment of non metastatic castration resistant prostate cancer (nmCRPC) who are at high risk of developing metastatic disease. in adult men for the treatment of metastatic hormone-sensitive prostate cancer (mHSPC) in combination with androgen deprivation therapy (ADT).
In an open-label, uncontrolled, multi-center, single-arm dedicated QT study in 45 patients with CRPC, an exposure-QT analysis suggested a concentration-dependent increase in QTcF for apalutamide and its active metabolite. Apalutamide demonstrated an antitumor activity in the mouse xenograft models of prostate cancer, where it decreased tumor cell proliferation and reduced tumor volume.
L02BB05
L - Antineoplastic and immunomodulating agents
L02 - Endocrine therapy
L02B - Hormone antagonists and related agents
L02BB - Anti-androgens
L02BB05 - Apalutamide
Absorption
Mean absolute oral bioavailability was approximately 100%. Median time to achieve peak plasma concentration (tmax) was 2 hours (range: 1 to 5 hours). Median tmax may be increased with a high-fat meal. Administration of oral apalutamide at recommended dosages resulted in a steady state within 4 weeks with a maximum peak concentration (Cmax) and AUC of 6.0 mcg/mL and 100 mcgh/mL, respectively. Cmax and AUC of apalutamide is expected to increase in a dose-proportional manner. The mean mean peak-to-trough ratio was 1.63 indicating low daily fluctuations in the plasma concentrations of the drug. The major active metabolite N-desmethyl apalutamide Cmax was 5.9 mcg/mL (1.0) and AUC was 124 mcgh/mL (23) at steady-state after the recommended dosage.
Route of Elimination
Apalutamide and its main active metabolite are subject to both renal and focal elimination. Up to 70 days following a single oral administration of radiolabeled apalutamide, 65% of the dose was recovered in urine (1.2% of dose as unchanged apalutamide and 2.7% as N-desmethyl apalutamide) and 24% was recovered in feces (1.5% of dose as unchanged apalutamide and 2% as N-desmethyl apalutamide).
Volume of Distribution
The mean apparent volume of distribution at steady-state of apalutamide was approximately 276 L.
Clearance
The CL/F of apalutamide was 1.3 L/h after single dosing and increased to 2.0 L/h at steady-state after once-daily dosing. An increase in apparent clearance (CL/F) was observed with repeat dosing, likely due to induction of apalutamides own metabolism. The auto-induction effect likely reached its maximum at the recommended dosage because exposure of apalutamide across the dose range of 30 to 480 mg is dose-proportional.
Apalutamide primarily undergoes CYP2C8 and CYP3A4-mediated metabolism to its pharmacologically active metabolite, N-desmethyl apalutamide. The contribution of CYP2C8 and CYP3A4 in the total metabolism of apalutamide is approximately 58% and and 13% following single dose but changes to 40% and 37%, respectively at steady-state. The auto-induction of CYP3A4-mediated metabolism by apalutamide may explain the increase in CYP3A4 enzymatic activity at steady-state. Based on systemic exposure, relative potency, and pharmacokinetic properties, N-desmethyl apalutamide likely contributed to the clinical activity of apalutamide.
The mean effective half-life for apalutamide in patients with NM-CRPC was approximately 3 days at steady-state.
Persistent androgen receptor (AR) signaling is a common feature of castration-resistant prostate cancer (CRPC), attributed to AR gene-amplification, AR gene mutation, increased AR expression or increased androgen biosynthesis in prostate tumors. Apalutamide is an antagonist of AR that to the binding-site in the ligand-binding domain of the receptor with the IC50 of 16 nM. Upon binding, apalutamide disrupts AR signalling, inhibits DNA binding, and impedes AR-mediated gene transcription. Apalutamide impairs the translocation of AR from the cytoplasm to the nucleus thus reduces the concentrations of AR available to interact with the androgen response-elements (AREs). Upon treatment with apalutamide, AR was not recruited to the DNA promoter-regions. Its main metabolite, N-desmethyl apalutamide, is a less potent inhibitor of AR, and exhibited one-third the activity of apalutamide in an in vitro transcriptional reporter assay.






