

X

API Suppliers

US DMFs Filed

CEP/COS Certifications
0

JDMFs Filed
Other Certificates
Other Suppliers
USA (Orange Book)
Europe

Canada

Australia

South Africa
Uploaded Dossiers
U.S. Medicaid
Annual Reports
0
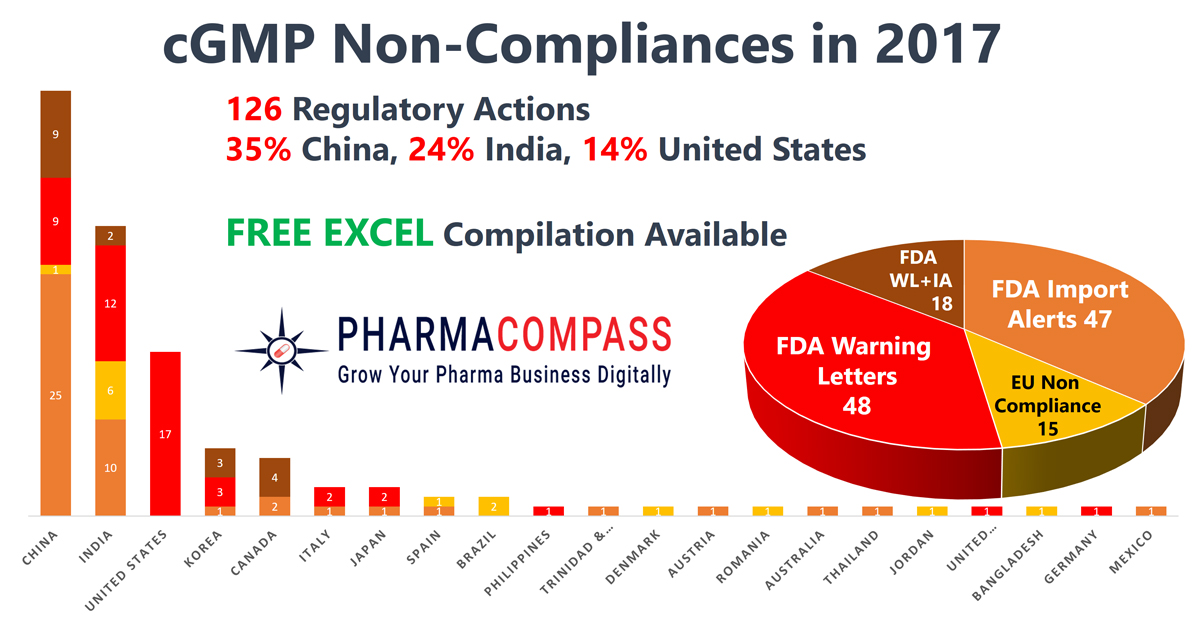
Impressions: 9113
https://www.pharmacompass.com/radio-compass-blog/2017-recap-of-warning-letters-import-alerts-and-non-compliances
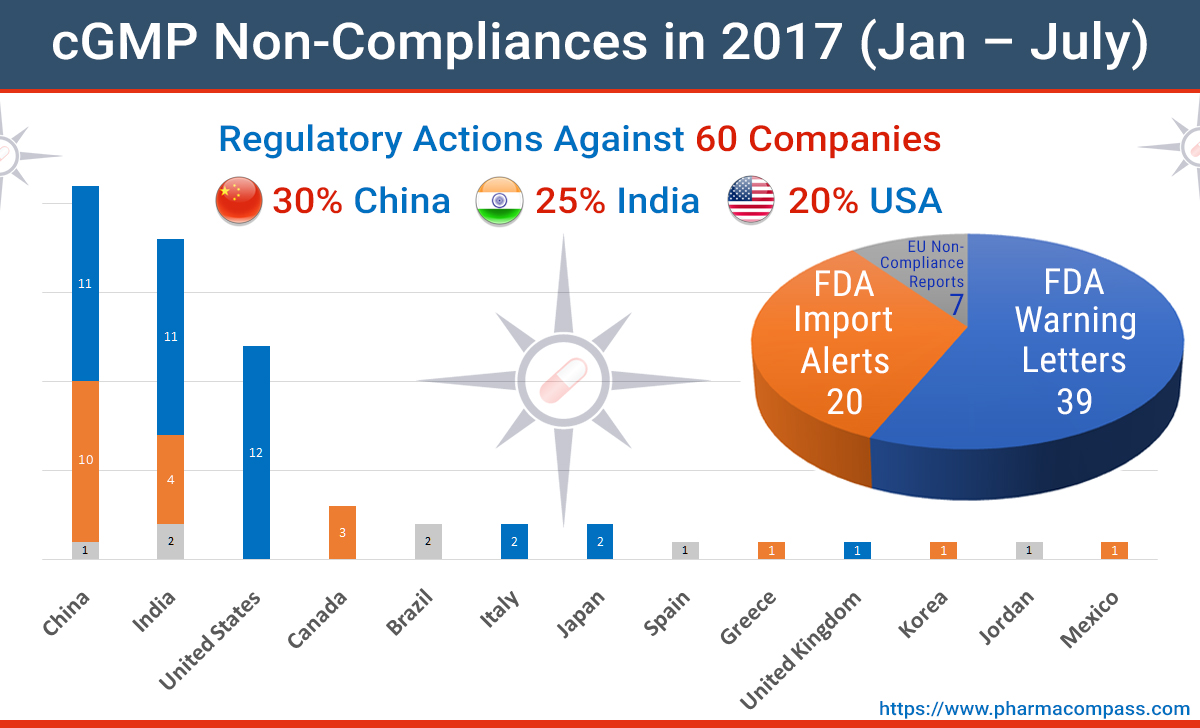
Impressions: 7732
https://www.pharmacompass.com/radio-compass-blog/mid-2017-recap-of-fda-warning-letters-import-alerts-eu-non-compliances

Impressions: 4004
https://www.pharmacompass.com/radio-compass-blog/fda-problems-at-pfizer-s-us-facility-continue-china-fda-joins-ich







