







Data
integrity continued to be a hot topic in the pharmaceutical industry through
2017. According to a recent analysis by GMP (good
manufacturing practices) intelligence expert, Barbara Unger, approximately 65
percent of all US Food and
Drug Administration (USFDA) warning letters
issued in FY2017 (October 1, 2016 until September 30, 2017) included a data integrity component.
However, in the previous year, this number was even higher — at 79 percent — implying there has been a decline in non-compliance incidents pertaining to data integrity in 2017.
Click here for our compilation of all non-compliances in 2017 (Excel version available) for FREE!
2017’s recurring concern – a failure
to thoroughly investigate problems
In PharmaCompass’ 2016 compilation, serious charges of blatant data manipulation surfaced at organizations around the world.
In 2017, we witnessed a reduction in data-integrity violations
uncovered at pharmaceutical manufacturers due to the absence of audit trail
software in quality control testing equipment.
However, the implementation of audit trails has resulted in the emergence of a new failing – the improper handling of out-of-specification (OOS) results.
Failure “to thoroughly investigate any unexplained discrepancy or failure of a batch” became a recurring theme in concerns highlighted at major generic players like Mylan, Fresenius, Teva, Dr Reddy’s, Hetero Labs and Lupin.
In the case of Fresenius’ oncology API plant in India, USFDA investigators found employees had halted and invalidated
HPLC (high-performance liquid chromatography) analyses nearly 250 times when they
believed the tests were going to end with OOS results.
The
USFDA warned Fresenius SE after the company’s Indian plant that makes cancer-drug ingredients for the US market aborted hundreds of drug-quality tests.
Click here for our compilation of all non-compliances in 2017 (Excel version available) for FREE!
The situation at Lupin wasn’t much better as the warning letter issued to its formulation manufacturing facilities in Goa and Indore (Pithampur Unit II) said the company failed to “thoroughly review any unexplained discrepancy” as Lupin invalidated approximately 96 percent of all OOS results obtained at Pithampur and over 75 percent of them in Goa.
Failure to resolve
recurring problems also led the USFDA to tell Meridian Medical
Technologies, a division of Pfizer that
makes the EpiPen injector
device (sold by Mylan NV), that serious component and product failures had
been associated with patient deaths.
In its warning
letter, the USFDA said the Pfizer unit failed to adequately investigate
problems at its manufacturing facility in Brentwood, Missouri. It also did not
take appropriate corrective actions before a USFDA inspection earlier this
year.
Meridian had received hundreds of complaints that
the EpiPen device, which is used to combat serious allergic reactions,
failed to operate during life-threatening emergencies.
Click here for our compilation of all non-compliances in 2017 (Excel version available) for FREE!
Non-compliances at
finished drug producers
outnumbered those at API facilities
During 2017, the
number of finished pharmaceutical companies cited for compliance concerns
significantly outnumbered the number of active pharmaceutical ingredient (API)
producers.
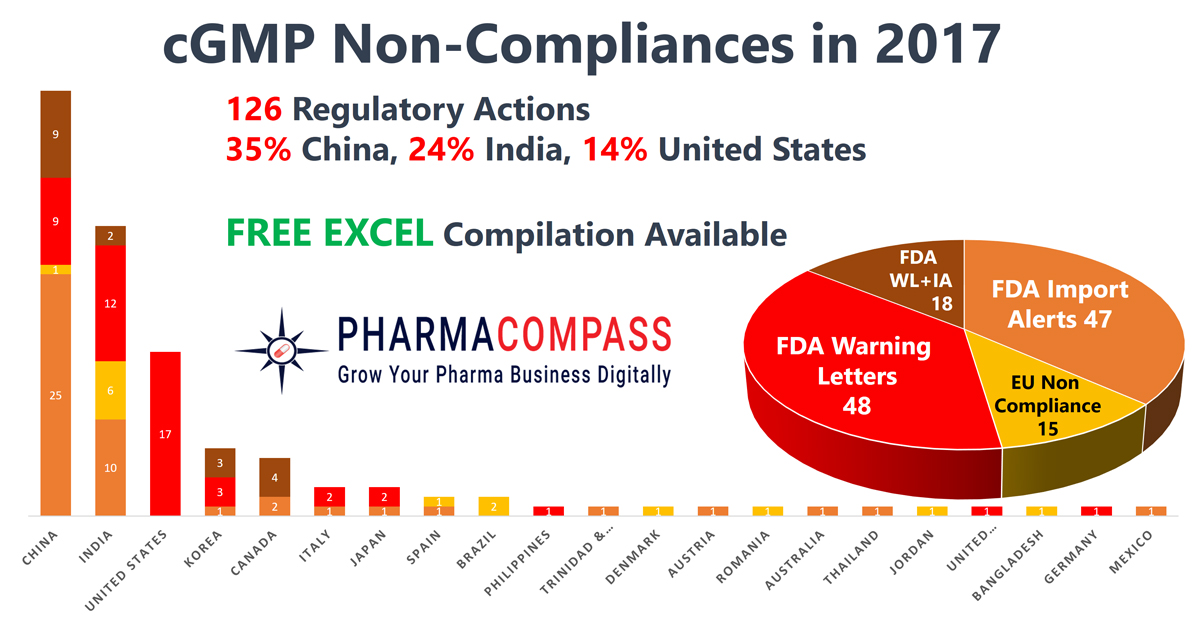
While the FDA issued 48 warning letters to drug product manufacturers, API producers received only 18 warning letters. The actions by the European regulators was similar — 15 non-compliance certificates were issued to finished drug producers, as compared to only two API manufactures who were found lagging behind in compliance standards.
China, India and the
United States continued to lead the countries where regulators found most
shortcomings.
As compared to the
previous year, in 2017 regulators issued fewer non-compliance certifications as
the FDA had
been hampered by staffing shortages. As a result, the FDA’s inspections in India “dropped 27 percent in fiscal 2017 from a year earlier, to 185 from 252.”
Click here for our compilation of all non-compliances in 2017 (Excel version available) for FREE!
In 2017, EU and
FDA started their mutual recognition program. But will it work?
2017 was also a
landmark year for the US and European regulators as the USFDA and the European Medicines Agency (EMA) announced their program
for mutual recognition of inspections of drug manufacturers, which
became operational on November 1, 2017.
The FDA will now recognize eight EU drug regulators – from Austria, Croatia, France, Italy, Malta, Spain, Sweden and the UK – as capable of conducting inspections of manufacturing facilities that meet the USFDA requirements.
This is an unprecedented move — prior to this, the USFDA had never recognized another country’s inspectorate.
As
part of the agreement, the European Commission (EC), the US FDA and the EMA signed a confidentiality commitment that allows the USFDA to share non-public
and commercially confidential information, including trade secret
information relating to inspections with European regulators.
As the mutual
recognition of inspections program goes live, there were examples of many
companies that were found to be consistently out of compliance by both the FDA
and regulators from the EU. Yet, there were cases where the regulators came to
different conclusions about the state of a particular facility they had
inspected.
While
European regulators had raised compliance concerns at Biocon, the company went ahead and got an FDA nod
for its biosimilar of Roche’s blockbuster cancer drug — Herceptin.
Click here for our compilation of all non-compliances in 2017 (Excel version available) for FREE!
The case of Qinhuangdao Zizhu — when WHO and FDA differed
In
another case, an inspection conducted by the USFDA at Qinhuangdao Zizhu Pharmaceutical from November 28 to December 1, 2016 uncovered
significant data integrity concerns and failures in the level of adherence to
current good manufacturing practices (cGMPs) for APIs.
In
the warning letter issued to the firm, the laboratory analysts admitted to the FDA inspectors that they had been “setting the clock back and repeating analyses for undocumented reasons.”
At
Qinhuangdao Zizhu, “initial sample results were overwritten or deleted” and the company “reported only the passing results from repeat analyses”.
In addition to not having effective measures to control data within their computerized systems, the FDA investigators found that the firm “relied on incomplete information” to determine whether Qinhuangdao Zizhu’s drugs met established specifications.
The investigators found “a recurring practice of re-testing samples until acceptable results were obtained” and that batch production records “contained blank or partially completed manufacturing data”.
On
March 8, 2017, Qinhuangdao Zizhu Pharmaceutical was placed on import alert
by the USFDA.
Click here for our compilation of all non-compliances in 2017 (Excel version available) for FREE!
Almost
a year prior to the USFDA inspection, in October 2015, the company had been
inspected by a WHO Prequalification Team (PQT) for levonorgestrel, mifepristone and ethinylestradiol APIs. The inspection found “five major deficiencies including data integrity issues and several minor deficiencies”.
The
WHO, however, went ahead and closed its inspection as compliant, based on
corrective and preventive actions (CAPAs) provided by the manufacturer.
In
view of the USFDA actions, and the fact that Qinhuangdao Zizhu Pharmaceutical
is the only WHO-PQT prequalified source of levonorgestrel API (as was seen in a similar case at Mylan), the WHO approach towards the compliance position was to
focus extensively on product quality.
READ: FDA and EU differ on cGMP standards at the same facilities: How will they mutually recognize inspections?
Our view
As the US regulators push hundreds of new generic drugs to market
in an effort to drive down prices of generic drugs in the United States, the
industry should get ready for an increasing number of inspections in the coming
years.
Our compilation indicates that in 2017, while most companies had installed the infrastructure necessary to combat issues related to data-integrity, there were problems that were systemic in nature. These ‘systemic problems’ remain, and the industry must get ready as the FDA and European inspectors join hands to crack down on them.
PharmaCompass’ 2017 Recap of FDA Warning Letters, Import Alerts & EU Non-Compliances is an easy way to evaluate companies that have run into compliance challenges so that appropriate risk mitigation strategies can be adopted.
Impressions: 9113
https://www.pharmacompass.com/radio-compass-blog/2017-recap-of-warning-letters-import-alerts-and-non-compliances
#PharmaFlow by PHARMACOMPASS
11 Jan 2018
Last year, data
integrity was a hot topic of discussion in the pharmaceutical industry.
According to a recent analysis by GMP (good manufacturing practices)
intelligence expert, Barbara Unger, approximately 80 percent of all FDA warning
letters in 2015 and 2016 included a data integrity component, and approximately
70 percent of the published European GMP non-compliance reports cited similar
shortcomings.
With a little over
half the year gone, PharmaCompass analyzed the regulatory action for
current GMP (cGMP)
non-compliance to evaluate how things are looking so far in 2017.
Click here to access the compilation of all 2017 non-compliances (Excel version available) for FREE!
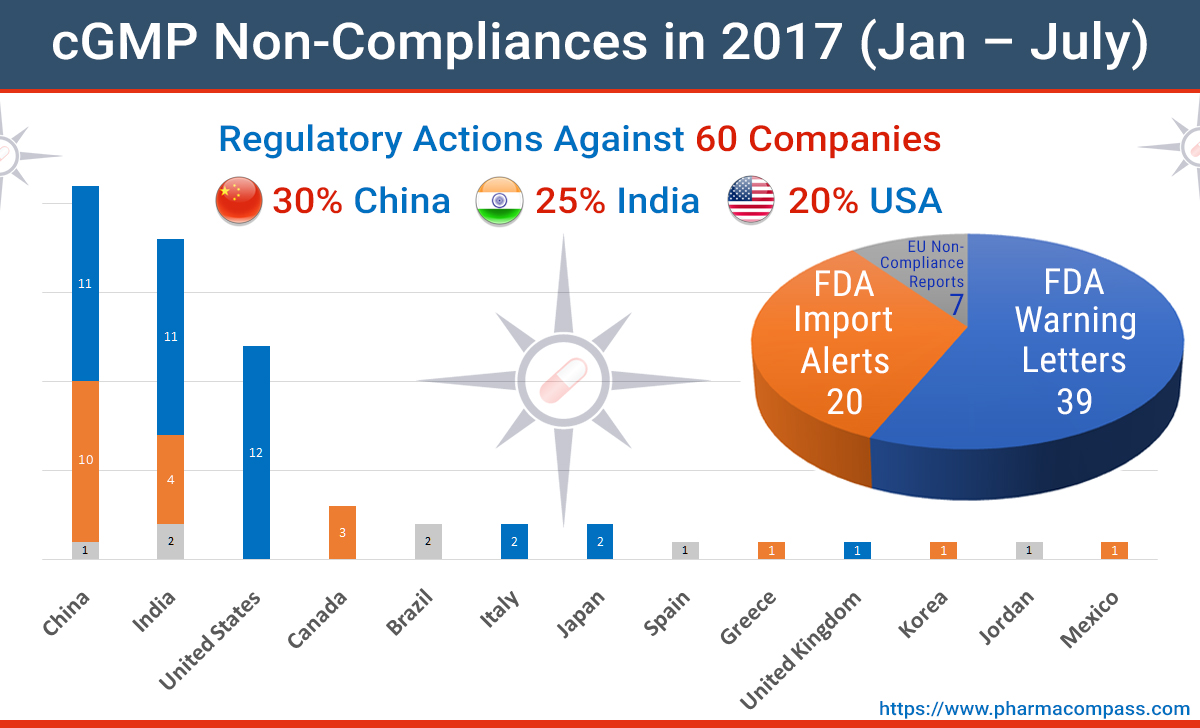
As
per our analysis, of all the non-compliance actions taken by the US and
European regulators, India and China continue to see the highest level of
activity, followed by the United States.
While most of the companies in the list are less known
pharmaceutical players, inspections uncovered deficiencies at leading companies
like Pfizer,
Teva,
Mylan
and B Braun.
Data integrity violations continue to remain high
The US Food
and Drug Administration (US FDA) and EU inspections continued to uncover data
integrity issues across countries such as India, China, Italy and Japan.
In a warning letter posted earlier this year, for a January 2016 inspection, FDA investigators uncovered data-integrity violations at ACS Dobfar’s Italian drug manufacturing facility — FACTA Farmaceutici SpA. At FACTA, for multiple lots of sterile drug product, the
original data showed failing results. However, the final data reportedly showed
passing results.
The company was found storing original data in an “unofficial” and uncontrolled electronic spreadsheet on a shared computer network drive. The analyst told investigators that original data was first recorded in the “unofficial” spreadsheet and later transcribed to an “official” form.
Investigators
also documented that employees at FACTA used paper shredders to destroy
critical laboratory and production records.
During an
inspection performed exactly at the same time, FACTA’s EU GMP certification was renewed by the Italian regulators!
Click here to access the compilation of all 2017 non-compliances (Excel version available) for FREE!
Discrepancies
in conclusions drawn by regulators
This year,
while the United States and the European Union (EU) finally announced that they will be able to utilize each other’s good manufacturing practice (GMP) inspections of pharmaceutical manufacturing facilities, the road ahead looks uncertain as, in addition to incidents like FACTA, there have been multiple instances of discrepancies in the conclusions arrived at by regulators.
This year saw
the WHO grant an all clear for a Mylan facility
where the FDA had data-integrity concerns and a
similar situation arose at an active pharmaceutical ingredient (API) facility
in China.
An
inspection conducted by the USFDA at Qinhuangdao Zizhu Pharmaceutical from November 28 to December 1, 2016
uncovered significant data integrity concerns and failures in the level of
adherence to cGMP for APIs.
Click here to access the compilation of all 2017 non-compliances (Excel version available) for FREE!
In
the warning letter issued to the firm, the laboratory analysts admitted to FDA inspectors that they had been “setting the clock back and repeating analyses for undocumented reasons.”
At Qinhuangdao Zizhu, “initial sample results were overwritten or deleted” and the company “reported only the passing results from repeat analyses”.
On
March 8, 2017, Qinhuangdao Zizhu Pharmaceutical was placed on import alert
by the USFDA.
Almost
a year prior to the USFDA inspection, in October 2015, the company had been
inspected by a WHO Prequalification Team (PQT) for levonorgestrel, mifepristone and ethinylestradiol APIs. The inspection concluded with “five major deficiencies, including data integrity issues and several minor deficiencies”.
The WHO went ahead and closed their inspection as ‘compliant’, based on corrective and preventive actions (CAPAs) provided by the manufacturer.
In view of the US FDA actions, and the fact that Qinhuangdao Zizhu
Pharmaceutical was the only WHO-PQT prequalified source of levonorgestrel API, as in the case of Mylan, the WHO approach towards the compliance position was focused extensively on product quality.
In Japan, at Sato Yakuhin Kogyo, FDA investigators found analysts
performed testing in duplicate without scientific justification, while in
China, the French Ministry of Health found manipulation, backdating and falsification
of GMP documents such as batch manufacturing records, GC (gas chromatography)
and HPLC (high performance liquid chromatographs) chromatograms at Chongqing
Succeway Pharmaceutical.
Click here to access the compilation of all 2017 non-compliances (Excel version available) for FREE!
Heparin makes
headlines again in China; Indian firm fakes strike
This year, concerns
over the testing of Heparin in China re-emerged when FDA issued a warning letter to a contract testing laboratory — Shandong Analysis and Test Center. However, the activities at these companies to deceive inspectors paled in comparison to Vikshara Trading in India where the company faked a strike to prevent an FDA inspection.
When the FDA
inspection finally occurred, the FDA obtained evidence that the firm actively
manufactured numerous products at the time of the supposed strike.
Concerns over
product quality in the United States
In a warning letter
issued to ChemRite CoPac in the US, the FDA found that the company was manufacturing oral drug
solutions using the same equipment that was being used to manufacture numerous
non-pharmaceutical materials including an industrial car care product. The car
care product being made was paraffin-based and carried labels such as “harmful or fatal if swallowed” and “keep out of reach of children.”
The ingredients of
these non-pharmaceutical products were extremely difficult to remove from the
manufacturing equipment, and could contaminate the drug products, the FDA said.
At Raritan Pharmaceuticals, a company that makes teething tablets, the FDA found the drug contained ingredients, such as belladonna, which could pose potential toxic effects for its consumers — infants and children under two years of age.
Click here to access the compilation of all 2017 non-compliances (Excel version available) for FREE!
Sterile drug
manufacturing continues to be a global challenge
Non-compliant
operations uncovered at Sato Pharmaceutical (Japan), Euro Far Allergi (Spain), Tubilux (Italy), Biocon (India), Nova DFL Industrie (Brazil), Pfizer’s US operations (ex-Hospira) show that aseptic sterile drug manufacturing continues to
be a global challenge as companies struggle to get into compliance.
In February
2015, Pfizer acquired a site in McPherson, Kansas, through its US$ 17 billion acquisition of Hospira. Pfizer was aware of Hospira's manufacturing record
when it struck the deal, as the company was issued FDA warning letters in four of the seven continents — Europe, North America, Asia and Australia.
Approval of two drugs have been held up this
year due to compliance concerns at McPherson.
Click here to access the compilation of all 2017 non-compliances (Excel version available) for FREE!
The USFDA also warned manufacturers of non-sterile, water-based drug products of Burkholderia cepacia
complex (BCC or B cepacia) contamination, as there were product recalls due to
this and other water-borne opportunistic pathogens found in pharmaceutical
water systems.
The regulator’s warning stemmed from a multi-state outbreak of infections. In March this year, Phispers had carried a news item on Badrivishal Chemicals & Pharmaceuticals,
a manufacturer of docusate sodium. It had been placed on FDA’s import alert list in December last year.
Concerns over
water systems were also mentioned in the warning letters issued to Humco
Holding Group in the United States and Resonance Laboratories in India.
Click here to access the compilation of all 2017 non-compliances (Excel version available) for FREE!
Omitting names of
original suppliers; shipping drugs from banned API makers
The USFDA
investigators found companies in India (Sal Pharma) and China (Suzhou Pharmaceutical Technology and Lumis Global Pharmaceuticals)
omitting the name and address of the original API
manufacturers on certificate of analysis and declared themselves to be the
manufacturers. In the case of Suzhou, one of its suppliers
was placed on FDA’s import alert list.
However, the company shipped API
manufactured from the banned supplier by providing misleading declarations.
Last year, PharmaCompass
had reported on Teva’s newly built sterile manufacturing facility in Godollo, Hungary, and the issues
highlighted by the FDA in its warning letter and the product recalls from this unit. As part of its global restructuring, while Teva is now winding up its sterile injectables plant in Godollo, it remains to be seen what decision
will be taken on its API manufacturing facility in Hangzhou (China), where the FDA had
highlighted concerns over process capability and the resulting impact on
product quality.
Wockhardt’s global compliance problems also continued with its manufacturing facility in the United States — Morton Grove Pharmaceuticals — receiving a warning letter.
Click here to access the compilation of all 2017 non-compliances (Excel version available) for FREE!
Our view
Earlier this year, a major Indian API manufacturer — Divi’s Laboratories — was placed on FDA’s import alert list. It was issued a warning letter due to a variety of problems which were uncovered at the site. The concerns raised at Divi’s along with other companies indicate a shifting focus on part of the FDA investigators from audit trails as there
is greater depth in the nature of the observations.
To assist the
industry, the FDA has started posting frequently requested Form 483s on a
routine basis which provides insight for the industry to track the new areas of
regulatory focus.
You can find the Form
483s on PharmaCompass at https://www.pharmacompass.com/radio-compass-news/Quality-Alerts
Click here to access the compilation of all 2017 non-compliances (Excel version available) for FREE!
Impressions: 7732
https://www.pharmacompass.com/radio-compass-blog/mid-2017-recap-of-fda-warning-letters-import-alerts-eu-non-compliances
#PharmaFlow by PHARMACOMPASS
10 Aug 2017

This week in our compliance round up, we look at the continuing problems at Pfizer’s McPherson unit in the US and China FDA’s entry into the ICH as a regulatory member. Yet again, WHO sources API from a plant in China, where USFDA had raised data integrity concerns. Meanwhile, Aurobindo Pharma’s Unit VII clears FDA inspection with zero observations.
Problems continue at Pfizer’s US facility; its sure-shot biosimilar gets rejected
Earlier this year,
Pfizer’s fill/finish manufacturing facility in McPherson, Kansas, received a warning letter from the US Food and Drug Administration (USFDA). The compliance concern was initially revealed in a press release issued by Momenta Pharmaceuticals — a biotechnology company developing a generic version of Teva’s long-acting Copaxone® 40mg/mL (glatiramer acetate injection). Momenta is working in
collaboration with Sandoz, which in turn has tied up with Pfizer as its fill/finish
manufacturing partner.
This week, a month after the FDA staff found that Pfizer’s biosimilar of Amgen’s drug Epogen (epoetin alfa) was nearly identical and an FDA advisory committee followed to recommend approval with a 14-1 vote, the FDA issued a complete response letter (CRL) to Pfizer.
The letter cited
concerns which had already been raised in the warning letter issued on February 14, 2017, following a
routine inspection of the facility.
This development
continues to be positive news for Teva, as Copaxone generated US$ 4.22 billion in sales last year. Continued
compliance concerns at Pfizer indicate that Momenta will still have to wait for
its generic to get approved.
While
previous FDA inspections at the McPherson facility had raised concerns over the assurance of product sterility, no warning letter had ever been issued to this site.
In
February 2015, Pfizer acquired the McPherson site through its US$ 17 billion acquisition of Hospira. Pfizer was aware of Hospira's manufacturing record
when it struck the deal, as the company was issued FDA warning letters in four of the seven continents — Europe, North America, Asia and Australia.
Once again, WHO sources API from Chinese firm where FDA had
raised concerns
Three
weeks after PharmaCompass shared the differences in assessments of the World Health Organization (WHO) and the USFDA over
the observations at a Mylan finished formulation
site in India, a similar situation has arisen at an active pharmaceutical
ingredient (API) facility in China.
An
inspection conducted by the USFDA at Qinhuangdao Zizhu Pharmaceutical from November 28 to December 1, 2016 uncovered significant data
integrity concerns and failures in the level of adherence to cGMP (current good
manufacturing practices) for APIs.
In
the warning letter issued to the firm, the laboratory analysts admitted to FDA inspectors that they had been “setting the clock back and repeating analyses for undocumented reasons.”
At Qinhuangdao Zizhu, “initial sample results were overwritten or deleted” and the company “reported only the passing results from repeat analyses”.
In addition to not having effective measures to control data within their computerized systems, the FDA investigators found that the firm “relied on incomplete information” to determine whether Qinhuangdao Zizhu’s drugs met established specifications.
The investigators found “a recurring practice of re-testing samples until acceptable results were obtained” and that batch production records “contained blank or partially completed manufacturing data”.
On
March 8, 2017, Qinhuangdao Zizhu Pharmaceutical was placed on the import alert by the USFDA.
Almost
a year prior to the USFDA inspection, in October 2015, the company had been
inspected by a WHO Prequalification Team (PQT) for levonorgestrel, mifepristone and ethinylestradiol APIs. The inspection concluded with “five major deficiencies including data integrity issues and several minor deficiencies”.
The
WHO went ahead and closed their inspection as compliant based on corrective and
preventive actions (CAPAs) provided by the manufacturer.
In
view of the USFDA actions, and the fact that Qinhuangdao Zizhu Pharmaceutical
is the only WHO-PQT prequalified source of levonorgestrel API, as in the case of Mylan, the WHO approach towards the compliance position has been
to focus extensively on product quality.
The WHO has requested manufacturers that use levonorgestrel manufactured by Qinhuangdao Zizhu to take “additional measures such as comprehensive testing upon receipt” to help ensure that the quality of all batches of levonorgestrel is assured.
In
addition, the WHO has said that procurement agencies may continue to procure
FPPs that contain API produced at Qinhuangdao Zizhu Pharmaceutical, until
further notice.
The WHO-PQT is planning to conduct an on-site inspection of
Qinhuangdao Zizhu Pharmaceutical and also plans to work with finished
pharmaceutical products manufacturers to identify additional sources for
levonorgestrel.
China
FDA approved as an ICH member
In a major boost to China’s pharmaceutical growth plans, the International Council for Harmonization (ICH) Assembly approved the China Food and Drug Administration
(CFDA) as a new Regulatory Member. This decision was taken during a meet in
Montreal, Canada, from May 31 to June 1, 2017.
While compliance concerns linger over some pharmaceutical factories in the country, China’s acceptance of this membership indicates that the country is prepared to bring its drug manufacturing and testing practices in line with international quality standards.
ICH was created in
1990 to bring regulatory authorities and the pharmaceutical industry together
in order to discuss scientific and technical aspects of drug registration. Over
the last 25 years, ICH has worked on achieving greater global harmonization so
that safe and effective drugs are developed and registered in the most
resource-efficient manner.
In India, Aurobindo’s Unit VII gets zero observations; Lupin founder passes away
Indian pharmaceutical industry lost a
veteran this week. Desh Bandhu Gupta, the founder chairman of Lupin and a self-made billionaire, died in Mumbai at the age of 79.
Five years back, he had handed over the reins of Lupin to his eldest daughter Vinita
Gupta and son Nilesh Gupta.
Desh Bandhu Gupta, or DBG as he was
known, was a deft businessman who founded Lupin in 1968 with just Rs 5,000
borrowed from his wife. Prior to this, he was the professor of chemistry at the
Birla Institute of Technology and Science, Pilani. He went on to become India’s 20th richest person with an estimated net worth of US$ 5.1 billion (according to the 2016 Forbes India Rich List).
While much of the industry abandoned making anti-TB drugs, given the low margins and the government's price controls, Lupin remained consistent as the world’s largest supplier of anti-TB drugs. DBG forged global alliances to develop new medicines to fight tuberculosis.
Meanwhile, another Indian company — Aurobindo Pharma — sailed through a USFDA inspection with zero observation. Aurobindo Pharma’s Unit VII is a formulations manufacturing facility and one of the
largest facilities for the company from which has filed the maximum ANDA
applications to the FDA.
According to a recent company presentation to investors, the company has filed 158 ANDAs (abbreviated new drug application) from this facility, of which 88 drugs received final approvals and 20 drugs have tentative approvals and 50 are currently under review.
Impressions: 4004
https://www.pharmacompass.com/radio-compass-blog/fda-problems-at-pfizer-s-us-facility-continue-china-fda-joins-ich
#PharmaFlow by PHARMACOMPASS
29 Jun 2017

