BLOG
MARKET INTEL by PharmaCompass
CONTENT by Suppliers
- Interview #SpeakPharma
- Video #SupplierSpotlight
- Vlog #PharmaReel
- Company Bio #AboutSupplier
- Services Bio #AboutCapabilities
News
Create content with us, ask us
Data integrity continued to be a hot topic in the pharmaceutical industry through 2017. According to a recent analysis by GMP (good manufacturing practices) intelligence expert, Barbara Unger, approximately 65 percent of all US Food and Drug Administration (USFDA) warning letters issued in FY2017 (October 1, 2016 until September 30, 2017) included a data integrity component.
However, in the previous year, this number was even higher — at 79 percent — implying there has been a decline in non-compliance incidents pertaining to data integrity in 2017.
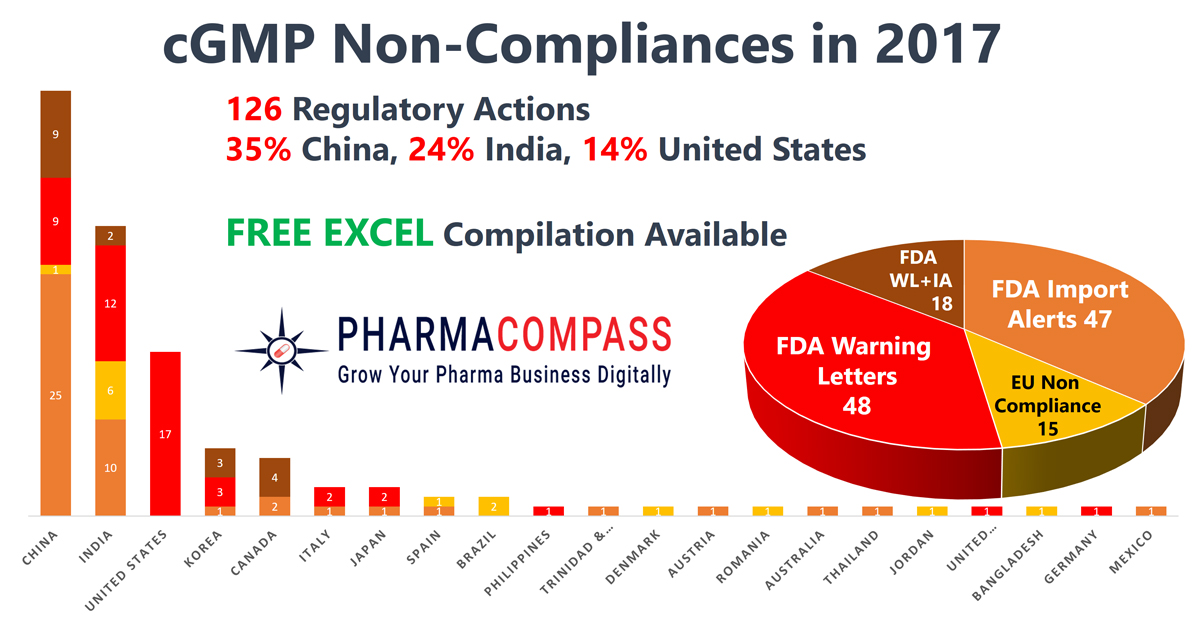
Click here for our compilation of all non-compliances in 2017 (Excel version available) for FREE!
2017’s recurring concern – a failure
to thoroughly investigate problems
In PharmaCompass’ 2016 compilation, serious charges of blatant data manipulation surfaced at organizations around the world.
In 2017, we witnessed a reduction in data-integrity violations uncovered at pharmaceutical manufacturers due to the absence of audit trail software in quality control testing equipment.
However, the implementation of audit trails has resulted in the emergence of a new failing – the improper handling of out-of-specification (OOS) results.
Failure “to thoroughly investigate any unexplained discrepancy or failure of a batch” became a recurring theme in concerns highlighted at major generic players like Mylan, Fresenius, Teva, Dr Reddy’s, Hetero Labs and Lupin.
In the case of Fresenius’ oncology API plant in India, USFDA investigators found employees had halted and invalidated HPLC (high-performance liquid chromatography) analyses nearly 250 times when they believed the tests were going to end with OOS results.
The USFDA warned Fresenius SE after the company’s Indian plant that makes cancer-drug ingredients for the US market aborted hundreds of drug-quality tests.
Click here for our compilation of all non-compliances in 2017 (Excel version available) for FREE!
The situation at Lupin wasn’t much better as the warning letter issued to its formulation manufacturing facilities in Goa and Indore (Pithampur Unit II) said the company failed to “thoroughly review any unexplained discrepancy” as Lupin invalidated approximately 96 percent of all OOS results obtained at Pithampur and over 75 percent of them in Goa.
Failure to resolve recurring problems also led the USFDA to tell Meridian Medical Technologies, a division of Pfizer that makes the EpiPen injector device (sold by Mylan NV), that serious component and product failures had been associated with patient deaths.
In its warning letter, the USFDA said the Pfizer unit failed to adequately investigate problems at its manufacturing facility in Brentwood, Missouri. It also did not take appropriate corrective actions before a USFDA inspection earlier this year.
Meridian had received hundreds of complaints that the EpiPen device, which is used to combat serious allergic reactions, failed to operate during life-threatening emergencies.
Click here for our compilation of all non-compliances in 2017 (Excel version available) for FREE!
Non-compliances at
finished drug producers
outnumbered those at API facilities
During 2017, the
number of finished pharmaceutical companies cited for compliance concerns
significantly outnumbered the number of active pharmaceutical ingredient (API)
producers.
While the FDA issued 48 warning letters to drug product manufacturers, API producers received only 18 warning letters. The actions by the European regulators was similar — 15 non-compliance certificates were issued to finished drug producers, as compared to only two API manufactures who were found lagging behind in compliance standards.
China, India and the United States continued to lead the countries where regulators found most shortcomings.
As compared to the previous year, in 2017 regulators issued fewer non-compliance certifications as the FDA had been hampered by staffing shortages. As a result, the FDA’s inspections in India “dropped 27 percent in fiscal 2017 from a year earlier, to 185 from 252.”
Click here for our compilation of all non-compliances in 2017 (Excel version available) for FREE!
In 2017, EU and
FDA started their mutual recognition program. But will it work?
2017 was also a landmark year for the US and European regulators as the USFDA and the European Medicines Agency (EMA) announced their program for mutual recognition of inspections of drug manufacturers, which became operational on November 1, 2017.
The FDA will now recognize eight EU drug regulators – from Austria, Croatia, France, Italy, Malta, Spain, Sweden and the UK – as capable of conducting inspections of manufacturing facilities that meet the USFDA requirements.
This is an unprecedented move — prior to this, the USFDA had never recognized another country’s inspectorate.
As part of the agreement, the European Commission (EC), the US FDA and the EMA signed a confidentiality commitment that allows the USFDA to share non-public and commercially confidential information, including trade secret information relating to inspections with European regulators.
As the mutual recognition of inspections program goes live, there were examples of many companies that were found to be consistently out of compliance by both the FDA and regulators from the EU. Yet, there were cases where the regulators came to different conclusions about the state of a particular facility they had inspected.
While European regulators had raised compliance concerns at Biocon, the company went ahead and got an FDA nod for its biosimilar of Roche’s blockbuster cancer drug — Herceptin.
Click here for our compilation of all non-compliances in 2017 (Excel version available) for FREE!
The case of Qinhuangdao Zizhu — when WHO and FDA differed
In another case, an inspection conducted by the USFDA at Qinhuangdao Zizhu Pharmaceutical from November 28 to December 1, 2016 uncovered significant data integrity concerns and failures in the level of adherence to current good manufacturing practices (cGMPs) for APIs.
In the warning letter issued to the firm, the laboratory analysts admitted to the FDA inspectors that they had been “setting the clock back and repeating analyses for undocumented reasons.”
At Qinhuangdao Zizhu, “initial sample results were overwritten or deleted” and the company “reported only the passing results from repeat analyses”.
In addition to not having effective measures to control data within their computerized systems, the FDA investigators found that the firm “relied on incomplete information” to determine whether Qinhuangdao Zizhu’s drugs met established specifications.
The investigators found “a recurring practice of re-testing samples until acceptable results were obtained” and that batch production records “contained blank or partially completed manufacturing data”.
On March 8, 2017, Qinhuangdao Zizhu Pharmaceutical was placed on import alert by the USFDA.
Click here for our compilation of all non-compliances in 2017 (Excel version available) for FREE!
Almost a year prior to the USFDA inspection, in October 2015, the company had been inspected by a WHO Prequalification Team (PQT) for levonorgestrel, mifepristone and ethinylestradiol APIs. The inspection found “five major deficiencies including data integrity issues and several minor deficiencies”.
The WHO, however, went ahead and closed its inspection as compliant, based on corrective and preventive actions (CAPAs) provided by the manufacturer.
In view of the USFDA actions, and the fact that Qinhuangdao Zizhu Pharmaceutical is the only WHO-PQT prequalified source of levonorgestrel API (as was seen in a similar case at Mylan), the WHO approach towards the compliance position was to focus extensively on product quality.
Our view
As the US regulators push hundreds of new generic drugs to market in an effort to drive down prices of generic drugs in the United States, the industry should get ready for an increasing number of inspections in the coming years.
Our compilation indicates that in 2017, while most companies had installed the infrastructure necessary to combat issues related to data-integrity, there were problems that were systemic in nature. These ‘systemic problems’ remain, and the industry must get ready as the FDA and European inspectors join hands to crack down on them.
PharmaCompass’ 2017 Recap of FDA Warning Letters, Import Alerts & EU Non-Compliances is an easy way to evaluate companies that have run into compliance challenges so that appropriate risk mitigation strategies can be adopted.
The PharmaCompass Newsletter – Sign Up, Stay Ahead
Feedback, help us to improve. Click here
Image Credit : cGMP Non Compliances in 2017 by PharmaCompass is licensed under CC BY 2.0
“ The article is based on the information available in public and which the author believes to be true. The author is not disseminating any information, which the author believes or knows, is confidential or in conflict with the privacy of any person. The views expressed or information supplied through this article is mere opinion and observation of the author. The author does not intend to defame, insult or, cause loss or damage to anyone, in any manner, through this article.”