

X

API Suppliers

US DMFs Filed
0

CEP/COS Certifications
0

JDMFs Filed
0
Other Certificates
0
Other Suppliers
0
USA (Orange Book)
Europe

Canada

Australia

South Africa
Uploaded Dossiers
0
U.S. Medicaid
Annual Reports
1. Plx 4032
2. Plx4032
3. R05185426
4. Rg 7204
5. Rg-7204
6. Rg7204
7. Zelboraf
1. 918504-65-1
2. Plx4032
3. Zelboraf
4. 1029872-54-5
5. Plx-4032
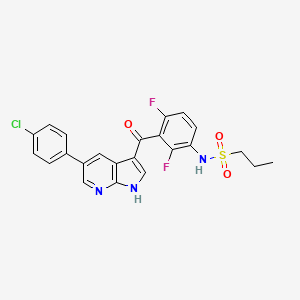
6. N-(3-(5-(4-chlorophenyl)-1h-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluorophenyl)propane-1-sulfonamide
7. Rg7204
8. Plx 4032
9. Rg 7204
10. Vemurafenib (plx4032, Rg7204)
11. N-(3-{[5-(4-chlorophenyl)-1h-pyrrolo[2,3-b]pyridin-3-yl]carbonyl}-2,4-difluorophenyl)propane-1-sulfonamide
12. Ro 5185426
13. Vemurafenib (plx4032)
14. Ro5185426
15. Rg-7204
16. 207smy3fqt
17. Ro-5185426
18. Chebi:63637
19. Mfcd18074504
20. Nsc761431
21. N-(3-((5-(4-chlorophenyl)-1h-pyrrolo(2,3-b)pyridin-3-yl)carbonyl)-2,4- Difluorophenyl)propane-1-sulfonamide
22. Vemurafenib;plx-4032
23. Ro-51-85426
24. 1-propanesulfonamide, N-(3-((5-(4-chlorophenyl)-1h-pyrrolo(2,3-b)pyridin-3- Yl)carbonyl)-2,4-difluorophenyl)-
25. 1-propanesulfonamide, N-[3-[[5-(4-chlorophenyl)-1h-pyrrolo[2,3-b]pyridin-3-yl]carbonyl]-2,4-difluorophenyl]-
26. N-[3-[[5-(4-chlorophenyl)-1h-pyrrolo[2,3-b]pyridin-3-yl]carbonyl]-2,4-difluorophenyl]-1-propanesulfonamide
27. N-[3-[5-(4-chlorophenyl)-1h-pyrrolo[2,3-b]pyridine-3-carbonyl]-2,4-difluorophenyl]propane-1-sulfonamide
28. N-{3-[5-(4-chlorophenyl)-1h-pyrrolo[2,3-b]pyridine-3-carbonyl]-2,4-difluorophenyl}propane-1-sulfonamide
29. Zelboraf (tn)
30. 1-propanesulfonamide, N-(3-((5-(4-chlorophenyl)-1h-pyrrolo(2,3-b)pyridin-3-yl)carbonyl)-2,4-difluorophenyl)-
31. 1415041-85-8
32. N-(3-((5-(4-chlorophenyl)-1h-pyrrolo(2,3-b)pyridin-3-yl)carbonyl)-2,4-difluorophenyl)-1-propanesulfonamide
33. Vemurafenib [usan]
34. Vemurafenib [usan:inn]
35. Unii-207smy3fqt
36. Vemurafenibum
37. Ro 51-85426
38. Hsdb 8143
39. N-(3-{(5-(4-chlorophenyl)-1h-pyrrolo(2,3-b)pyridin-3-yl)carbonyl}-2,4- Difluorophenyl)propane-1-sulfonamide
40. 3og7
41. Vemurafenib [mi]
42. Vemurafenib; Plx4032
43. Vemurafenib [inn]
44. Vemurafenib [jan]
45. Plx4032 - Vemurafenib
46. Vemurafenib [vandf]
47. Vemurafenib [mart.]
48. Vemurafenib [who-dd]
49. Schembl298931
50. Vemurafenib (jan/usan/inn)
51. Gtpl5893
52. Chembl1229517
53. Dtxsid50238710
54. Ex-a053
55. Vemurafenib [orange Book]
56. Hms3265m03
57. Hms3265m04
58. Hms3265n03
59. Hms3265n04
60. Hms3654p09
61. Hms3748g15
62. Bcp25783
63. Ex-a1335
64. Bdbm50396483
65. Nsc800964
66. S1267
67. Zinc52509366
68. Akos007930804
69. Am81259
70. Ccg-264883
71. Cs-0216
72. Db08881
73. Me-0096
74. Nsc-761431
75. Nsc-800964
76. Pb11741
77. Ncgc00250399-01
78. Ncgc00250399-05
79. Ncgc00250399-08
80. Vemurafenib, Rg7204, Ro5185426
81. Ac-25010
82. Hy-12057
83. N-[3-[5-(4-chlorophenyl)-1h-pyrrolo[2,3-b]pyridine-3-carbonyl]-2,4-difluoro-phenyl]propane-1-sulfonamide
84. Sy067868
85. Ft-0660388
86. Ft-0675792
87. Ft-0689782
88. Sw218095-2
89. A25476
90. A25742
91. D09996
92. R-7204
93. Ab01273970-01
94. Ab01273970_03
95. Q423111
96. Sr-01000941568
97. Carbonyl]-2,4-difluorophenyl}propane-1-sulfonamide
98. J-522975
99. J-690009
100. Sr-01000941568-1
101. Brd-k56343971-001-02-3
102. Brd-k56343971-001-05-6
103. Plx4032,vemurafenib, Rg7204, Ro5185426, Zelboraf
104. N-{3-[5-(4-chlorophenyl)-1h-pyrrolo[2,3-b]pyridine-3-
105. [(2s,5r)-2,5-dimethyl-4-[(tetrahydro-2h-pyran-4-yl)methyl]-1-piperazinyl][3-[(5-fluoro-2-methyl-4-pyrimidinyl)amino]-4,6-dihydro-6,6 -dimethylpyrrolo[3,4-c]pyrazol-5(1h)-yl]methanone
106. N-(3-(5-(4-chlorophenyl)-1h-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluorophenyl)-propane-1-sulfonamide
107. Propane-1-sulfonic Acid {3-[5-(4-chloro-phenyl)-1h-pyrrolo[2,3-b]pyridine-3-carbonyl]-2,4-difluoro-phenyl}-amide
| Molecular Weight | 489.9 g/mol |
|---|---|
| Molecular Formula | C23H18ClF2N3O3S |
| XLogP3 | 5 |
| Hydrogen Bond Donor Count | 2 |
| Hydrogen Bond Acceptor Count | 7 |
| Rotatable Bond Count | 7 |
| Exact Mass | 489.0725466 g/mol |
| Monoisotopic Mass | 489.0725466 g/mol |
| Topological Polar Surface Area | 100 Ų |
| Heavy Atom Count | 33 |
| Formal Charge | 0 |
| Complexity | 790 |
| Isotope Atom Count | 0 |
| Defined Atom Stereocenter Count | 0 |
| Undefined Atom Stereocenter Count | 0 |
| Defined Bond Stereocenter Count | 0 |
| Undefined Bond Stereocenter Count | 0 |
| Covalently Bonded Unit Count | 1 |
| 1 of 2 | |
|---|---|
| Drug Name | Zelboraf |
| PubMed Health | Vemurafenib (By mouth) |
| Drug Classes | Antineoplastic Agent |
| Drug Label | ZELBORAF (vemurafenib) is a kinase inhibitor available as 240 mg tablets for oral use. Vemurafenib has the chemical name propane-1-sulfonic acid {3-[5-(4-chlorophenyl)-1H-pyrrolo[2,3-b]pyridine-3-carbonyl]-2,4-difluoro-phenyl}-amide. It has the molec... |
| Active Ingredient | Vemurafenib |
| Dosage Form | Tablet |
| Route | Oral |
| Strength | 240mg |
| Market Status | Prescription |
| Company | Hoffmann La Roche |
| 2 of 2 | |
|---|---|
| Drug Name | Zelboraf |
| PubMed Health | Vemurafenib (By mouth) |
| Drug Classes | Antineoplastic Agent |
| Drug Label | ZELBORAF (vemurafenib) is a kinase inhibitor available as 240 mg tablets for oral use. Vemurafenib has the chemical name propane-1-sulfonic acid {3-[5-(4-chlorophenyl)-1H-pyrrolo[2,3-b]pyridine-3-carbonyl]-2,4-difluoro-phenyl}-amide. It has the molec... |
| Active Ingredient | Vemurafenib |
| Dosage Form | Tablet |
| Route | Oral |
| Strength | 240mg |
| Market Status | Prescription |
| Company | Hoffmann La Roche |
Vemurafenib is used for the treatment of unresectable or metastatic melanoma with BRAF V600E mutation. Vemurafenib is designated an orphan drug by the US Food and Drug Administration (FDA) for the treatment of this cancer. An FDA-approved diagnostic test (e.g., cobas 4800 BRAF V600 Mutation Test) is required to confirm the presence of the BRAF V600E mutation prior to initiation of therapy. /Included in US product label/
American Society of Health-System Pharmacists 2013; Drug Information 2013. Bethesda, MD. 2013, p. 1233
Zelboraf is not recommended for use in patients with wild-type BRAF melanoma.
US Natl Inst Health; DailyMed. Current Medication Information for ZELBORAF (vemurafenib) tablet, film coated (April 2012). Available from, as of July 17, 2013: https://dailymed.nlm.nih.gov/dailymed/lookup.cfm?setid=38eea320-7e0c-485a-bc30-98c3c45e2763
Serious hypersensitivity reactions (e.g., anaphylaxis, generalized rash and erythema, hypotension) have been reported in patients receiving vemurafenib. Vemurafenib should be permanently discontinued in patients who experience a severe hypersensitivity reaction.
American Society of Health-System Pharmacists 2013; Drug Information 2013. Bethesda, MD. 2013, p. 1234
Photosensitivity reactions (mild to severe) have been reported in 33-49% of patients receiving vemurafenib in clinical trials. If intolerable grade 2 (i.e., tender erythema covering 10-30% of body surface area) or greater reaction occurs, the dosage of vemurafenib should be reduced.
American Society of Health-System Pharmacists 2013; Drug Information 2013. Bethesda, MD. 2013, p. 1234
Vemurafenib prolongs the QT interval in a concentration-dependent manner. In a multicenter, open-label, phase 2 study, QT interval prolongation was evaluated in patients with BRAF V600E mutation-positive, metastatic melanoma who were receiving vemurafenib (960 mg twice daily). A maximum mean corrected QT (QTc) interval change from baseline of 12.8 msec during the first month of treatment and 15.1 msec during the first 6 months of treatment was observed in these patients. The manufacturer does not recommend initiation of vemurafenib in patients with electrolyte abnormalities unresponsive to corrective measures or congenital long QT syndrome. In addition, concomitant use of vemurafenib with drugs known to prolong the QT interval (e.g., class Ia and III antiarrhythmic agents) is not recommended. ECGs and serum electrolyte concentrations, including concentrations of potassium, magnesium, and calcium, should be obtained prior to initiation of therapy or following dosage modification, and monitored 15 days following initiation of therapy, then monthly for the first 3 months of treatment, and then every 3 months thereafter or more often as clinically indicated. Interruption or discontinuance of vemurafenib may be necessary if increases in the QTc interval occur during therapy with the drug.
American Society of Health-System Pharmacists 2013; Drug Information 2013. Bethesda, MD. 2013, p. 1234
Severe skin reactions (e.g., Stevens-Johnson syndrome, toxic epidermal necrolysis) have been reported with vemurafenib. If severe skin reactions occur, vemurafenib therapy should be permanently discontinued.
American Society of Health-System Pharmacists 2013; Drug Information 2013. Bethesda, MD. 2013, p. 1234
For more Drug Warnings (Complete) data for Vemurafenib (18 total), please visit the HSDB record page.
Vemurafenib is approved since 2011 for the treatment of metastatic melanoma with a mutation on BRAF in the valine located in the exon 15 at codon 600, this mutation is denominated as V600E. The V600E mutation, a substitution of glutamic acid for valine, accounts for 54% of the cases of cutaneous melanoma. Vemurafenib approval was extended in 2017, for its use as a treatment of adult patients with Erdheim-Chester Disease whose cancer cells present BRAF V600 mutation. Erdheim-Chester disease is an extremely rare histiocyte cell disorder that affects large bones, large vessels, central nervous system, as well as, skin and lungs. It is reported an association of Erdheim-Chester disease and V600E mutation.
FDA Label
Vemurafenib is indicated in monotherapy for the treatment of adult patients with BRAF-V600-mutation-positive unresectable or metastatic melanoma.
BRAF activation results in cell growth, proliferation, and metastasis. BRAF is an intermediary molecule in MAPK whose activation depends on ERK activation, elevation of cyclin D1 and cellular proliferation. The mutation V600E produces a constitutively form of BRAF. Vemurafenib has been shown to reduce all activation markers related to BRAF; in clinical trials, vemurafenib treatment showed a reduction of cytoplasmic phosphorylated ERK and a cell proliferation driven by Ki-67. Studies also reported decrease in MAPK-related metabolic activity. All the different reports indicate thet Vemurafenib generates an almost complete inhibition of the MAPK pathway.
Antineoplastic Agents
Substances that inhibit or prevent the proliferation of NEOPLASMS. (See all compounds classified as Antineoplastic Agents.)
Protein Kinase Inhibitors
Agents that inhibit PROTEIN KINASES. (See all compounds classified as Protein Kinase Inhibitors.)
L01EC01
L01XE15
S76 | LUXPHARMA | Pharmaceuticals Marketed in Luxembourg | Pharmaceuticals marketed in Luxembourg, as published by d'Gesondheetskeess (CNS, la caisse nationale de sante, www.cns.lu), mapped by name to structures using CompTox by R. Singh et al. (in prep.). List downloaded from https://cns.public.lu/en/legislations/textes-coordonnes/liste-med-comm.html. Dataset DOI:10.5281/zenodo.4587355
L - Antineoplastic and immunomodulating agents
L01 - Antineoplastic agents
L01E - Protein kinase inhibitors
L01EC - B-raf serine-threonine kinase (braf) inhibitors
L01EC01 - Vemurafenib
Absorption
Vemurafenib is well absorbed after oral administration. Peak concentrations are reached in 3 hours when an oral dose of 960 mg twice daily for 15 days has been given to patients. In the same conditions, Vemurafenib presents a Cmax of 62 mcg/ml and AUC of 601 mcg h/ml. It is unknown how food affects the absorption of vemurafenib. It presents an accumulation ratio of 7.36 after repeating doses of 960 mg
Route of Elimination
Analysis showed that 94% of administered Vemurafenib is excreted via feces and 1% is excreted by urine.
Volume of Distribution
The estimation of the volume of distribution for Vemurafenib is 106 L.
Clearance
The total body clearance is 31 L/day.
Following oral administration of (14)C-vemurafenib 960 mg in the tablet formulation, plasma samples were analyzed over 48 hours for vemurafenib and its metabolites. Mean data showed that vemurafenib and its metabolites represented 95% and 5% of the components in plasma, respectively.
US Natl Inst Health; DailyMed. Current Medication Information for ZELBORAF (vemurafenib) tablet, film coated (April 2012). Available from, as of July 17, 2013: https://dailymed.nlm.nih.gov/dailymed/lookup.cfm?setid=38eea320-7e0c-485a-bc30-98c3c45e2763
Vemurafenib is highly bound (> 99%) to human albumin and alpha-1 acid glycoprotein plasma proteins. The population apparent volume of distribution for vemurafenib in metastatic melanoma patients is estimated to be 106 L (with 66% inter-patient variability).
US Natl Inst Health; DailyMed. Current Medication Information for ZELBORAF (vemurafenib) tablet, film coated (April 2012). Available from, as of July 17, 2013: https://dailymed.nlm.nih.gov/dailymed/lookup.cfm?setid=38eea320-7e0c-485a-bc30-98c3c45e2763
The bioavailability of vemurafenib has not been determined. Following oral administration of vemurafenib at 960 mg twice daily for 15 days to patients with metastatic melanoma, the median Tmax was approximately 3 hours. Following 15 days of dosing at 960 mg twice daily, the mean (+ or - SD) Cmax and AUC0-12 were 62 ug/mL + or - 17 and 601 + or - 170 ug*h/mL, respectively. The median accumulation ratio estimate from the population pharmacokinetic analysis for the twice daily regimen is 7.36, with steady state achieved at approximately 15 to 22 days following dosing at 960 mg twice daily. At steady state, the mean vemurafenib exposure in plasma is stable (concentrations before and 2-4 hours after the morning dose) as indicated by the mean ratio of 1.13. The potential effect of food on vemurafenib absorption has not been studied. In clinical trials, vemurafenib was administered without regard to food.
US Natl Inst Health; DailyMed. Current Medication Information for ZELBORAF (vemurafenib) tablet, film coated (April 2012). Available from, as of July 17, 2013: https://dailymed.nlm.nih.gov/dailymed/lookup.cfm?setid=38eea320-7e0c-485a-bc30-98c3c45e2763
Following oral administration of (14)C-vemurafenib 960 mg in the tablet formulation, approximately 94% of the radioactive dose was recovered in feces and approximately 1% was recovered in the urine. The population apparent clearance of vemurafenib in patients with metastatic melanoma is estimated to be 31 L/day (with 32% inter-patient variability).
US Natl Inst Health; DailyMed. Current Medication Information for ZELBORAF (vemurafenib) tablet, film coated (April 2012). Available from, as of July 17, 2013: https://dailymed.nlm.nih.gov/dailymed/lookup.cfm?setid=38eea320-7e0c-485a-bc30-98c3c45e2763
For more Absorption, Distribution and Excretion (Complete) data for Vemurafenib (6 total), please visit the HSDB record page.
Vemurafenib is metabolized by CYP3A4 and the metabolites make up 5% of the components in plasma. The parent compound makes up for the remaining 95%.
The results from in vitro studies indicate that CYP3A4 was the major enzyme responsible in the metabolism of vemurafenib. The formation of mono-hydroxyl metabolites were inhibited for approximately 82% using the CYP inhibitor ketoconazole. No significant inhibition in the metabolism was observed in human liver microsomes in the presence of quinidine (CYP2D6 inhibitor), sulfaphenazole (CYP2C9 inhibitor), tranylcypromine (CYP2A6 inhibitor) and (-)-N-3-benzyl-phenobarbital (CYP2C19 inhibitor). In addition, CYP3A4 was responsible for the formation of the mono-hydroxylation metabolites.
European Medicines Agency (EMA), Committee for Medicinal Products for Human Use (CHMP), European Public Assessment Report (EPAR): Vemurafenib p.20 (2012). Available from, as of July 17, 2013: https://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002409/WC500124400.pdf
In vitro metabolism was analyzed for rat, mouse, dog, cynomolgus and human. The metabolism of vemurafenib was investigated both in vitro using microsomes and hepatocytes of various species and in vivo in rat, dog and human. In vitro analysis of vemurafenib metabolism in liver hepatocytes at the concentration of 10 uM, humans, dogs, and cynomolgus monkeys did not metabolize vemurafenib extensively (unchanged vemurafenib > or = 89%).
European Medicines Agency (EMA), Committee for Medicinal Products for Human Use (CHMP), European Public Assessment Report (EPAR): Vemurafenib p.19 (2012). Available from, as of July 17, 2013: https://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002409/WC500124400.pdf
In study /of patients/, identification of vemurafenib and metabolites in plasma, feces and urine was made for the first 96 hr, with a total collection period of 432 hrs (18 days). Mean data from the 7 patients indicated that over the period investigated (0 to 96 hours), potential metabolites each accounted for < 0.5% of the total administered dose in urine and .6% of the total administered dose in feces. In pooled fecal samples up to 48 hours post post-dose, parent compound accounted for at least 94% of total radioactivity (37% of the dose). In fecal samples taken 48-96 hr post-dose, the amount of metabolites increased, with M6, M3, and M8 representing approximately 19%, 14% and 12%, of the total chromatographic peak area, respectively (mean values) or 3%, 5% and 4% of the dose, respectively. Over the 0-96 hr collection period, potential metabolites M3 (mono-hydroxy) and M6 (glucosylation) each accounted for <0.5% of the total administered dose in urine. Vemurafenib accounted for approximately 1% of the total dose in urine.
European Medicines Agency (EMA), Committee for Medicinal Products for Human Use (CHMP), European Public Assessment Report (EPAR): Vemurafenib p.33 (2012). Available from, as of July 17, 2013: https://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002409/WC500124400.pdf
The elimination half-life of Vemurafenib is estimated to be 57 hours (range of 30-120 hours).
Single dose studies to determine pharmacokinetics were conducted in mouse, rat, rabbit, dog and monkey. In all pre-clinical species, half-lifes were between 2 and 5 hours ... . Only after intraperitoneal (IP) administration in mice, the half-life was much longer (20.6 h). Compared with other species, rabbits showed higher plasma exposure levels with a longer mean terminal half-life between 12 and 18 hours.
European Medicines Agency (EMA), Committee for Medicinal Products for Human Use (CHMP), European Public Assessment Report (EPAR): Vemurafenib p.19 (2012). Available from, as of July 17, 2013: https://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002409/WC500124400.pdf
The median of the individual elimination half-life estimates for vemurafenib is 57 hours (the 5th and 95th percentile range is 30 to 120 hours).
US Natl Inst Health; DailyMed. Current Medication Information for ZELBORAF (vemurafenib) tablet, film coated (April 2012). Available from, as of July 17, 2013: https://dailymed.nlm.nih.gov/dailymed/lookup.cfm?setid=38eea320-7e0c-485a-bc30-98c3c45e2763
Vemurafenib is an orally available inhibitor of mutated BRAF-serine-threonine kinase. Vemurafenif is a small molecule that interacts as a competitive inhibitor of the mutated species of BRAF. It is especially potent against the BRAF V600E mutation. Vemurafenib blocks downstream processes to inhibit tumour growth and eventually trigger apoptosis. Vemurafenib does not have antitumour effects against melanoma cell lines with the wild-type BRAF mutation.
Cutaneous squamous cell carcinoma (cuSCC) has been reported in patients with metastatic melanoma and CRC treated with vemurafenib. Clinical findings indicate that cuSCC may be related to treatment with vemurafenib. In order to understand the potential mechanism by which vemurafenib treatment contributes to development of cuSCC, vemurafenib was tested in vivo in the A431 cuSCC xenograft model. There was dose-dependent tumour growth stimulation of the xenograft tumours at doses higher than 25 mg/kg bid. The optimal dose of 75 mg/kg bid of vemurafenib caused a 103% induction of growth compared to the control (p=0.002). Immunohistochemistry showed staining of pERK only in the tumour samples treated with vemurafenib (75 mg/kg) as compared to the vehicle treated control group. Combination studies of vemurafenib and a MEK inhibitor, RO5068760, were performed to confirm inhibition of pERK.
European Medicines Agency (EMA), Committee for Medicinal Products for Human Use (CHMP), European Public Assessment Report (EPAR): Vemurafenib p.17 (2012). Available from, as of July 17, 2013: https://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002409/WC500124400.pdf
The effect of vemurafenib on RAF-MEK-ERK pathway inhibition was investigated in a panel of cancer cell lines, including melanoma cell lines expressing BRAFV600E, BRAFV600D, BRAFV600R, or BRAFWT. MEK and ERK phosphorylation (pMEK and pERK respectively) immunoassays were conducted to measure the levels of pMEK and pERK in various cancer cells treated with vemurafenib compared to vehicle control In cells expressing mutated BRAF (Colo829,WM2664 and WM1341D), vemurafenib inhibited both pERK and pMEK in a dose dependent manner. However, cells expressing BRAF WT vemurafenib induced rather than inhibited ERK or MEK phosphorylation in the cells expressing BRAFWT, such as HCT116, CHL-1 and SK-MEL-2 cells.
European Medicines Agency (EMA), Committee for Medicinal Products for Human Use (CHMP), European Public Assessment Report (EPAR): Vemurafenib p.14 (2012). Available from, as of July 17, 2013: https://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002409/WC500124400.pdf
Vemurafenib is a low molecular weight, orally available, inhibitor of some mutated forms of BRAF serine-threonine kinase, including BRAFV600E. Vemurafenib also inhibits other kinases in vitro such as CRAF, ARAF, wild-type BRAF, SRMS, ACK1, MAP4K5 and FGR at similar concentrations. Some mutations in the BRAF gene including V600E result in constitutively activated BRAF proteins, which can cause cell proliferation in the absence of growth factors that would normally be required for proliferation. Vemurafenib has anti-tumor effects in cellular and animal models of melanomas with mutated BRAFV600E.
US Natl Inst Health; DailyMed. Current Medication Information for ZELBORAF (vemurafenib) tablet, film coated (April 2012). Available from, as of July 17, 2013: https://dailymed.nlm.nih.gov/dailymed/lookup.cfm?setid=38eea320-7e0c-485a-bc30-98c3c45e2763







