


FDA’s December 2025 OPOE list features 784 prescription drugs, 73 OTC drugs
This
week, PharmaCompass brings you key highlights of the US Food and Drug Administration’s D
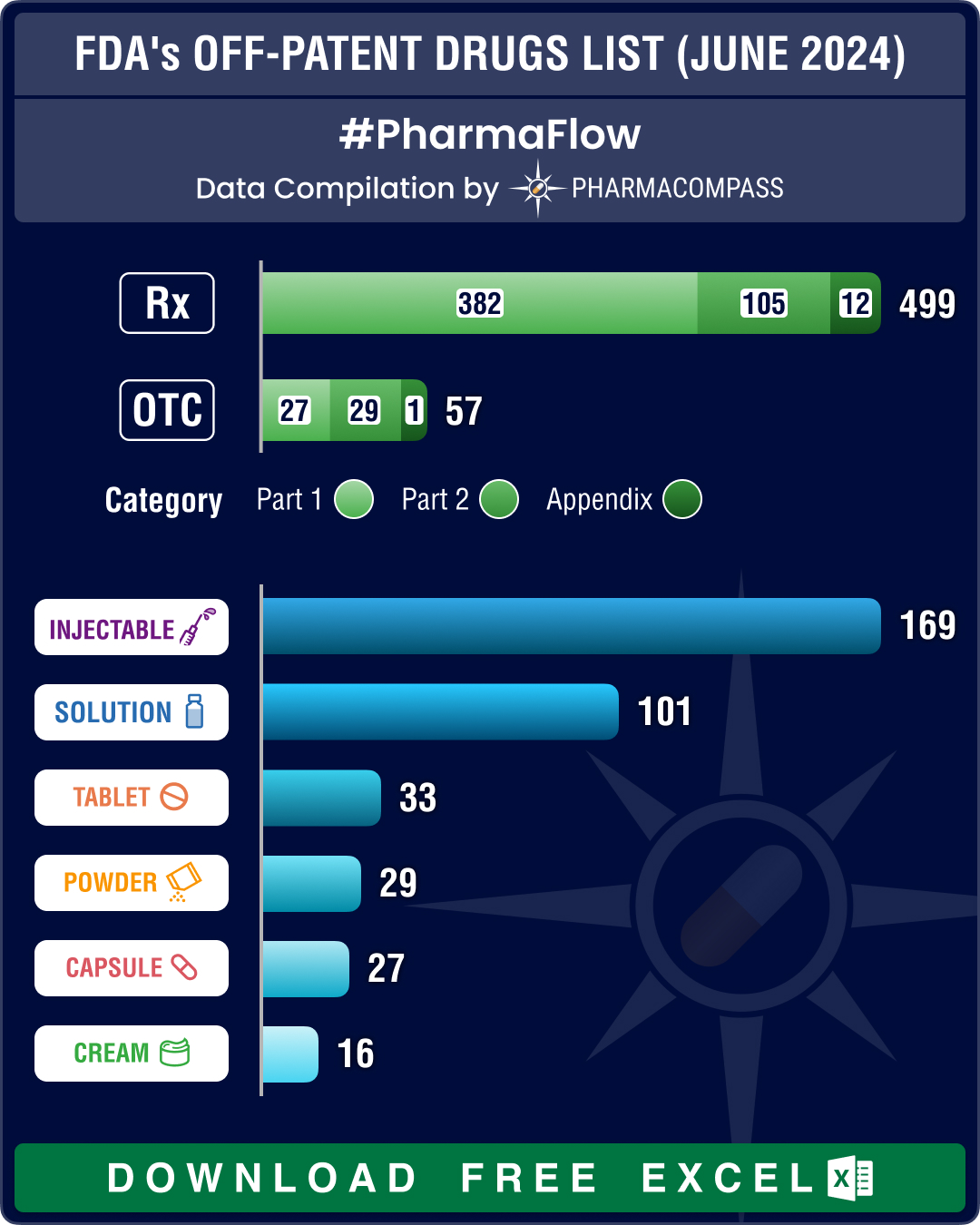
FDA’s June 2024 list of off-patent, off-exclusivity drugs sees rise in cancer, HIV treatments
This week PharmaCompass brings to you key highlights of the US Food and Drug Administration’s
- Privacy policy
- Terms and conditions
- Disclaimers
-

- Product listings are provided for informational purposes only. We do not supply or sell any products. Any products that may be covered by patent(s) are supplied solely for uses permitted under Section 107A of the Indian Patents Act and not for commercial sale.